GV6 Features at a Glance What’s New in GV6 Prices
- Intro. &
Examples - Building
Molecs. - Calc.
Setup - Large
Molecs. - Conform.
Searching - Plots &
Spectra - Surfaces &
Contours - Animations
& Movies
Contours
& Movies
GaussView 6 is the most advanced and powerful graphical interface available for Gaussian 16. With GaussView, you can build or import the molecular structures that interest you, set up, launch, monitor and control Gaussian calculations, and view the predicted results graphically, all without ever leaving the application. GaussView 6 includes many new features designed to make working with large systems of chemical interest convenient and straightforward. It also provides full support for all of the new modeling methods and capabilities available in Gaussian 16.
This brief introduction is a Quick Start to using GaussView 6 to investigate molecules and reactions with Gaussian 16. We invite you to try the techniques described here with your own molecules.
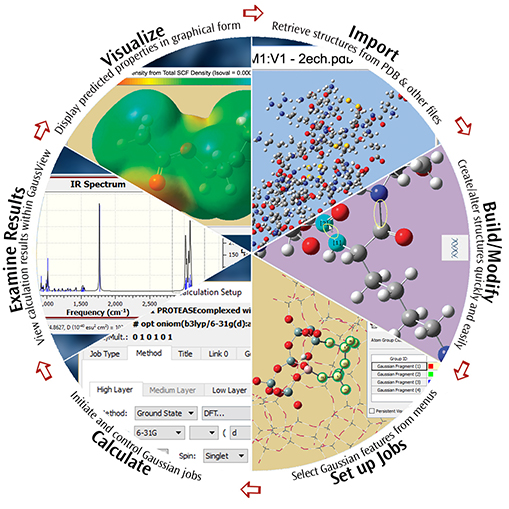
Examples: Visualizing Molecules & Reactions with GaussView 6
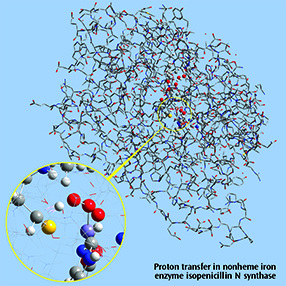 |
Close up: Proton transfer IRC, nonheme iron enzyme isopenicillin N synthase (IPNS)This 5368-atom system was studied with the ONIOM method in Gaussian, and the results were visualized in GaussView 6. For illustration clarity, hydrogen atoms in the low layer are omitted from display in both the close up and full molecule views. The ONIOM high accuracy layer is visualized in ball-and-stick format; the low accuracy layer is visualized in wire frame format in the close up view and in tube format in the whole molecule view [Lundberg09]. |
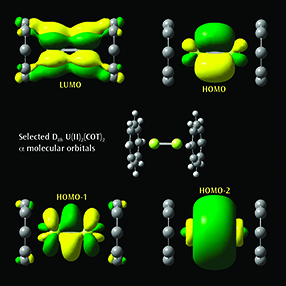
Selected α molecular orbitals for U(II)2(COT)2Each monomer has 4 U valence electrons available for metal-metal bonding: 2 electrons in f σ-type MOs and 2 unpaired electrons in f δ-type MOs. Beginning at the upper left and moving clockwise, the MOs visualized in GaussView 6 are the LUMO, HOMO, and the second-lowest and next-lowest energy MOs below the HOMO; all have D8h symmetry [Zhou10]. |
 |
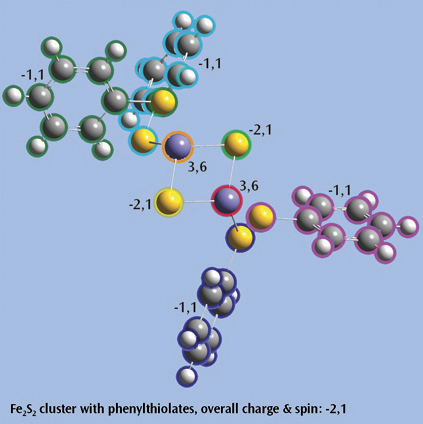 |
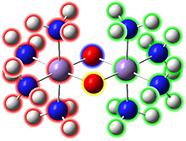
Fe2S2 cluster with phenylthiolatesThis is an open shell singlet system with charge -2. It has been set up for a Gaussian fragment guess calculation to model antiferromagnetic coupling. Each iron atom and bridging sulfur atom is placed in its own fragment, and each phenylthiolate group similarly defines a fragment, resulting in a total of eight fragments. GaussView 6 will automatically place the individual charge and spin multiplicity values for the eight fragments (labeled in the illustration) into the route section of the Gaussian job. The resulting wavefunction is stable and optimizes to a proper minimum. |
Building Molecules: Easier Than Ever
GaussView provides powerful molecule building features, the most important of which are listed below.
 |
|
Demonstration: Building Phenylpyridine
We will demonstrate several of these bulding features by building phenylpyridine: 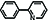
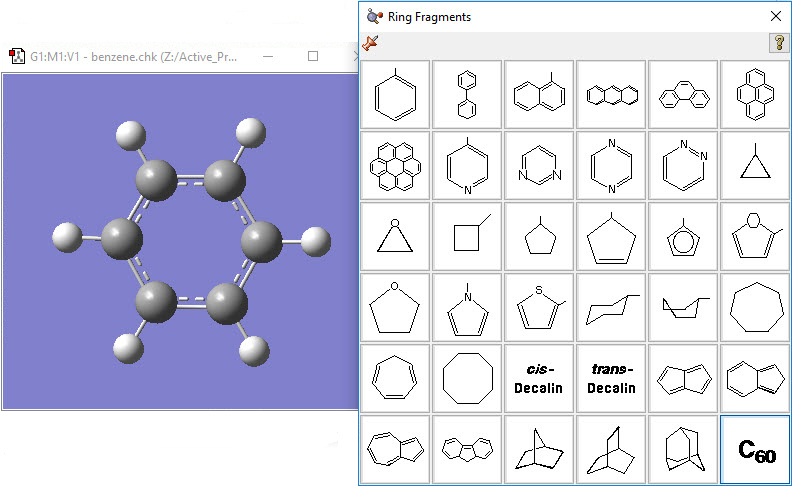 |
1: Select benzene from the Ring Fragment palette, and click inside the View window to add the ring. |
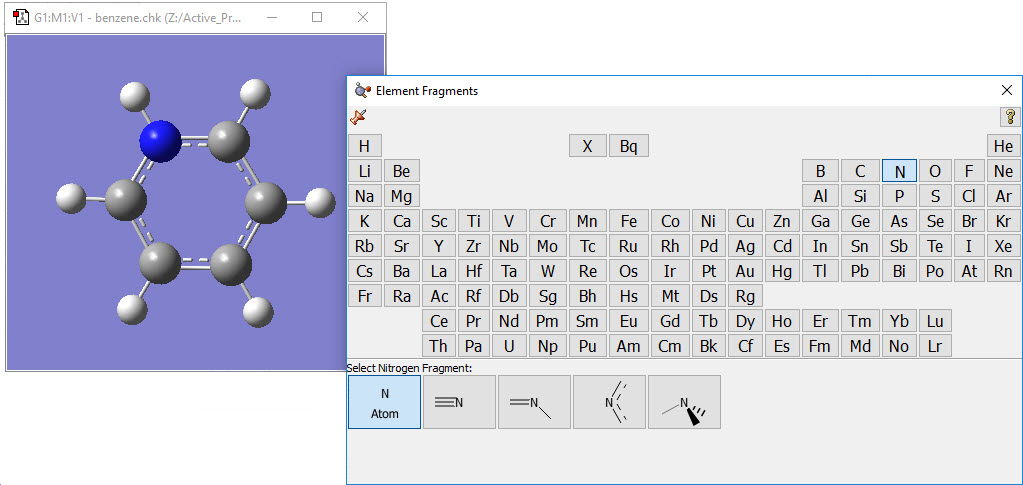 |
2: Use the Element Fragment palette to select nitrogen, and replace one of the carbon atoms. |
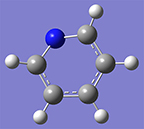 |
3: Use the Delete atom tool to remove the hydrogen atom attached to the nitrogen atom. Once you have completed this, hit the Rebond button and then the Clean button. The structure will regularize. |
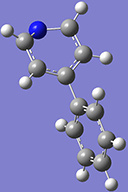 |
4: Return to the Ring Fragment panel, and select the benzene ring again. Click on the hydrogen atom attached to the carbon atom opposite the nitrogen atom to add the second ring. |
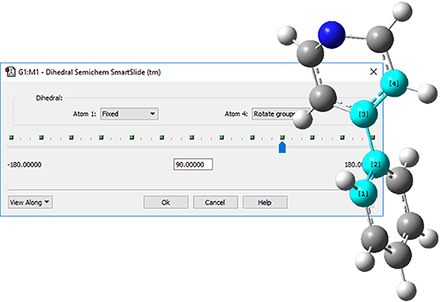 |
5: The angle between the two rings is wrong, so select the Modify Dihedral icon to twist it back. Click on the indicated atoms, and make sure that Rotate Groups is selected for both Atom 1 and Atom 4. Change the value in the angle text box to 90 degrees. Phenylpyridine is now complete. |
The following illustrations depict other advanced building features offered by GaussView 6.
Constraining Molecular Symmetry
 |
We have enabled point group symmetry constraints for this D0h bridged iron complex. When we click on the hydrogen atom above the left iron atom (with tetrahedral carbon as the current builder fragment), a methyl group is automatically added to all four symmetry-equivalent hydrogen atoms. |
Mirroring and Inverting Symmetry
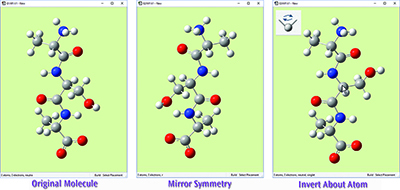
The original molecule is on the left. The middle view shows this tripeptide species after using Mirror Symmetry. The view on the right shows the result of using Invert About Atom by clicking on the central carbon atom (indicated with the cursor).
Studying Periodic Systems
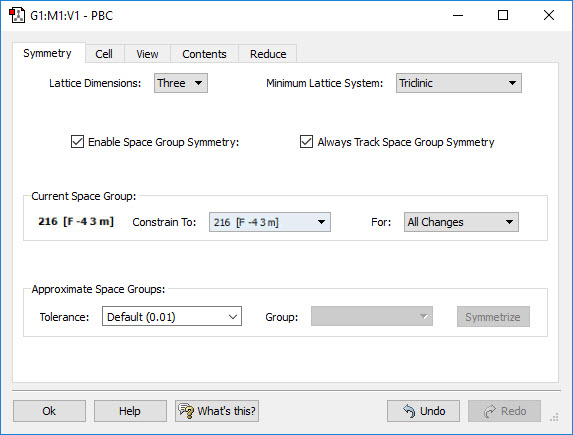
Gaussian 16 can perform Periodic Boundary Conditions (PBC) calculations to model periodic systems in condensed phases: i.e., polymers, surfaces and crystals. GaussView 6 provides a powerful facility for building such systems and generating their molecule specifications. The dialog to the left illustrates the space group symmetry capabilities; the space group for diamond has been selected for this 3D periodic structure.
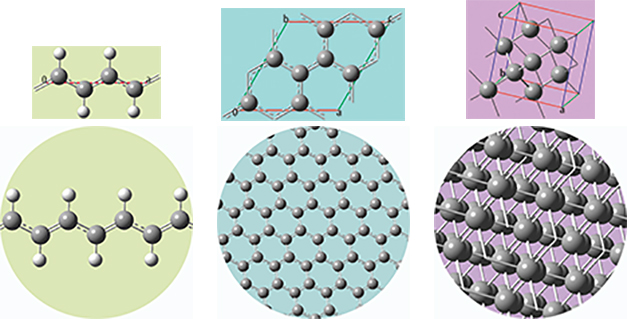
The unit cells for 1D, 2D and 3D periodic structures based on carbon are shown here, along with displays of multiple cells for the trans-polyvinyl acetate polymer, graphite sheet, and diamond crystal that they model. GaussView 6 will set up a PBC calculation, automatically including the translation vector(s) within the molecule specification.
GaussView 6’s Gaussian Calculation Setup dialog provides a menu-based interface to Gaussian 16 keywords and options. It allows you to set up virtually all types of Gaussian calculations, and it can submit from them as well.
Context-Aware Menus & Options
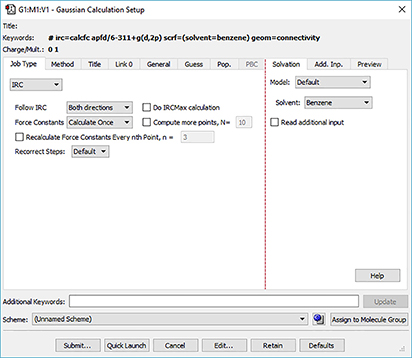
GaussView 6 automatically displays related settings for whatever selections you make in the Gaussian Calculation Setup dialog, and its various tabs provide access to all major Gaussian 16 features. In the dialog below, we are setting up an IRC calculation. Options for the direction to follow (forward and/or reverse), for obtaining the initial force constants and for specifying other IRC features are made available on the Job Type panel (left side of the figure). Similarly, once we have requested a calculation in solution, a menu with the available solvents appear on the Solvation panel (see the right side of the illustration).
Special Features for Specific Job Types
GaussView 6 makes it easy to set up Gaussian calculations that require additional specifications beyond keywords and options by providing graphic features for doing so, including the following:
- Assign atoms to ONIOM layers in several ways (as we saw). GaussView 6 also makes it easy to specify molecular mechanics atom types and charges.
- Reorder and repopulate MOs for CASSCF and other jobs.
- Add and redefine redundant internal coordinates.
- Specify frozen atoms/coordinates during geometry optimizations.
- Set atom equivalences for QST2 and QST3 transition state optimizations.
- Define fragments for fragment guess/counterpoise calculations, including assigning fragment-specific charges and spin multiplicities.
- Select atoms for normal mode analysis (frequency calculations).
- Specify atoms for NMR spin-spin coupling.
- Build unit cells for polymers, 2D surfaces and crystals for use in periodic boundary conditions (PBC) calculations.
- Preview the Gaussian Input file before it is submitted.
Demonstration: Setting up a QST3 Optimization
Setting up a QST3 optimization is made simple by several GaussView features, as we will demonstrate here. We are modeling the SN2 reaction:
C2O2H6 + H2O···OH–→ C1H3COO– + CH3OH + H2O
We want to locate the transition structure. Setting up the TS optimization in GaussView 6 is very easy.
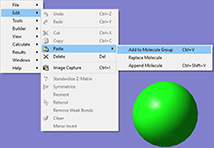
1: First, we build the reactant. We then copy the structure into a second frame within the same molecule group. This results in a second molecule with identical atom ordering, as required by Opt=QST3.
2: We go on to transform that structure into the product. We move the CH3 group bond from methyl acetate to the OH- group, modifying the orientation of its hydrogen atoms.
3: We copy the reactants again as the third member of the molecule group in order to set up the transition state guess structure. We modify the structure to conform to the TS guess we want to specify.
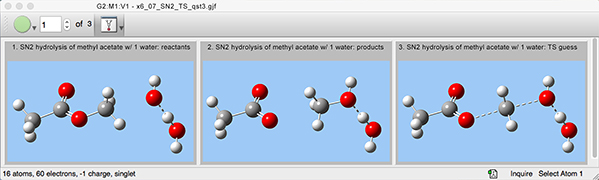
4: We can now specify the job details with the Gaussian Calculation Setup dialog. We also indicate that the TS guess is molecule 3 within the molecule group.
The input file for this calculation is now complete and ready to run.
The preceding example illustrates another Gaussian 16 convenience feature of GaussView 6: calculation schemes. Selecting an item from the Scheme menu causes a saved set of Gaussian keywords and options to be applied to the current job:
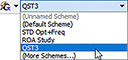
Using schemes in conjunction with the Gaussian Quick Launch feature allows you to initiate a calculation on the current molecule with a single click.
Specifying Alternate Isotopes
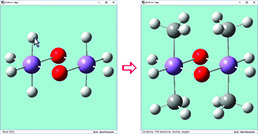
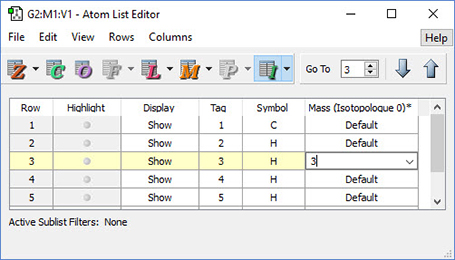
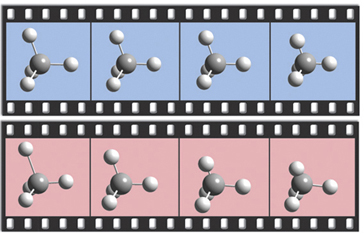
The dialog below shows GaussView 6’s facility for defining isotopes for use in subsequent frequency analyses (including multiple isotopologues if needed). In this example, two isotopologues are defined: one with the standard (most abundant) isotopes, and a second with tritium substituted for one of the hydrogen atoms. The animations illustrate the resulting change in the symmetric H stretch normal mode; the tritium-substituted atom is the one with the elongated motion in the frames with the rose background.
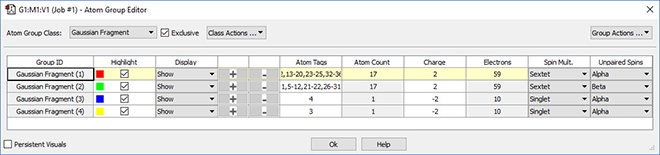
Defining Fragments
GaussView 6 makes it easy to define fragments for use in Gaussian 16 counterpoise, fragment guess and related calculations. We use the Atom Group Editor to define fragments and set fragment-specific charges and spin multiplicities in order to model antiferromagnetic coupling.
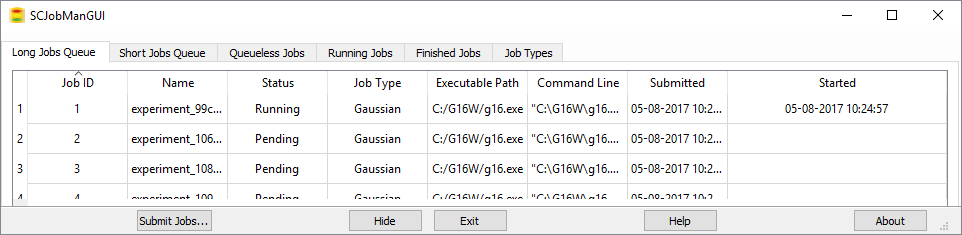
Managing Jobs
GaussView 6’s integrated Job Manager makes it simple to run and monitor jobs via a built-in queuing system. You can easily organize your processes within the three included queues. Just a click on the Running Jobs or Finished Jobs tab gives you the status of all of your calculations. You can also customize various job types by setting a default queue and other specifications.
GaussView 6 provides comprehensive support for importing and working with structures from PDB files:
- Select the desired structure(s) from multi-structure files.
- Add hydrogen atoms to all atoms automatically or manually according to user preference.
- Selectively add hydrogen atoms to one or more residues, chains, helices or other defined structural entities.
- Highlight/select atoms in individual residues or secondary structures.
- Quickly determine residue membership for any atom selected with the mouse.
- Easily assign atoms to ONIOM layers based on a variety of flexible criteria.
- Retain residue information within Gaussian 16 calculations and retrieved Gaussian 16 results.
The Gaussian 16 ONIOM method is typically used to study large molecules of biological interest. GaussView 6 contains several methods for assigning atoms to the distinct layers used by ONIOM to divide the problem into regions of differing modeling accuracy/computational cost.
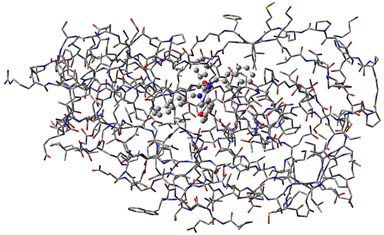 |
1: This molecule is HIV protease (Type 1) complexed with an inhibitor. The majority of the molecule is displayed as a wire frame, and the inhibitor is displayed in ball-and-stick format, indicating their status as the high accuracy and low accuracy regions (respectively) in an ONIOM calculation. We imported the structure from a PDB files. |
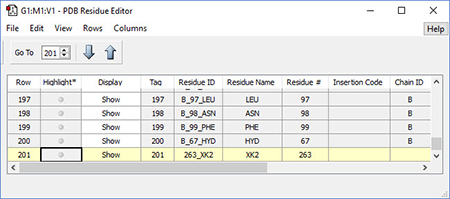 |
2: The PDB Residue Editor provides a convenient way to interact with large molecules on a per-residue basis. Here we have sorted the residues by name and are now working with the atoms in the inhibitor. We will go on to use other tools to assign atoms to ONIOM layers. |
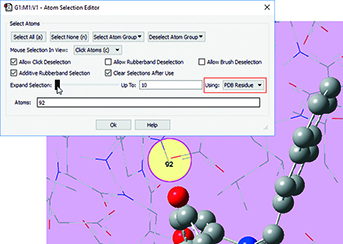 |
3: We want to add residues in close proximity to the inhibitor to the high layer. First, we zoom in on the relevant part of the complex, and select a nearby atom with the mouse. Using the Atom Selection Editor’s Expand Selection slider, we can expand the selection to include the entire residue containing that atom. |
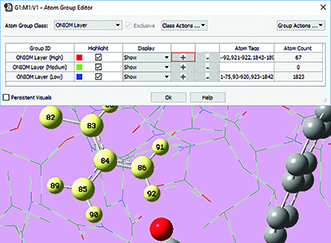 |
4: Once the desired atoms are selected, we use the Atom Group Editor to quickly add them to the desired ONIOM layer by clicking on the plus sign button in the corresponding row. We can continue to select and add nearby residues in this way until the specification of the high layer is complete. |
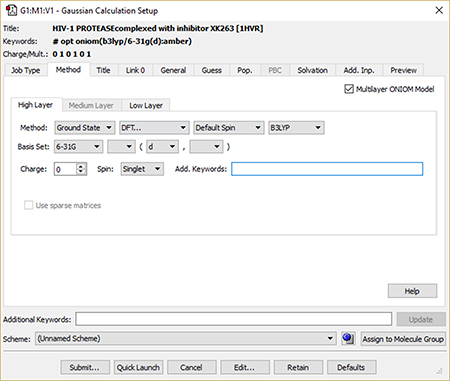 |
5: We set up and run a Gaussian 16 calculation using the Gaussian Calculation Setup dialog. We check the box selecting an ONIOM calculation and use the per-layer components of the Method panel to select the desired model chemistry for each layer. When we are ready, clicking the Submit button will initiate the Gaussian 16 calculation. |
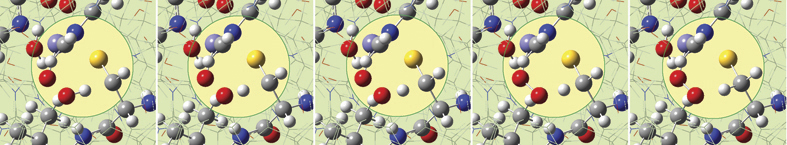
6: We eventually locate a transition structure and run an IRC calculation for a proton transfer reaction involving this system. GaussView 6 can display plots and animations from such studies. The five frames above depict structures from the reaction path. The corresponding points on the energy plot below have been marked with asterisks. We suppressed the display of low layer atoms in the highlighted region and added spotlights for clarity.
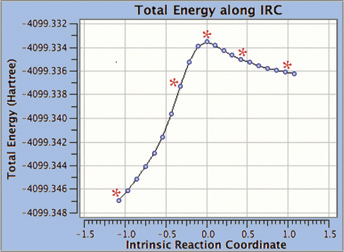
The optional GMMX module for GaussView 6 enables you to perform a conformational search. In addition, you can set up follow-on calculations for all the structures the search locates in a single step. We will demonstrate this process here.
We have already built methyl-β-D-glucose, seen here.

1: We want to use GaussView 6 to search for conformations for methyl-β-D-glucose, so we navigate to the GMMX Conformer dialog and specify the parameters for the GMMX Module. For this example, we want to find as many conformations as possible. We select Both for Search Method, and then select all the bonds and rings, allowing them to break when searching for conformations. We also increase the default energy range from 3.5 kcal/mol to 15 kcal/mol.
2: Once the job has been submitted and it completes, a dialog will pop up informing you that the calculation has completed. At this point, you choose which of the located conformations to open; we opted to open all of them. A new molecule group containing the selected structures will open.
Our search found 96 conformations. The multi-view window format for a molecule group is often convenient when working with multiple molecular structures, as seen at the right.
3: We are now ready to set up our follow-on calculations. We specify an APFD/6-311G+(2d,p) optimization. By clicking the Assign to Molecule Group button, we apply these settings to every molecule in the molecule group.
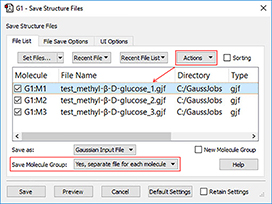
4: We will now save individual input files for the follow-on jobs. We select Yes, separate files for each molecule from the Save Molecule Group pop-up menu in the Save Structure Files dialog. This allows us to save each molecule to a separate file in a single operation. We add a molecule number to each file name using an item on the Actions menu.
5: Once we have saved the files, we run these jobs. After they complete, we identify the lowest energy unique conformations. We then set up and run calculations to predict their VCD spectra.
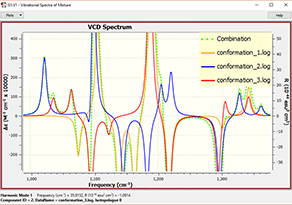
6: In order to view the predicted VCD spectrum, we begin by loading all of the VCD calculation results files into a single molecule group. We display the VCD results and then use the plot Mixture Editor (dialog on the right below) to produce a composite Boltzmann-averaged spectrum. The plot on the left shows the composite results (green dotted line) as well as those for the three most abundant conformations. We can easily see that conformation 1 dominates (orange line).
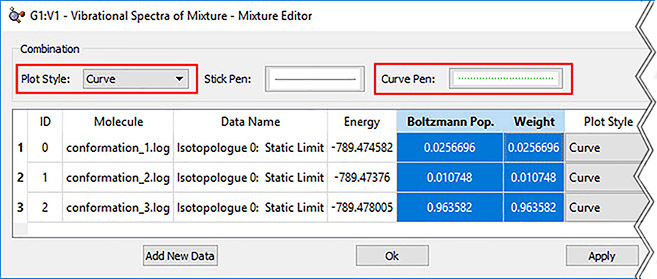
GaussView 6 presents predicted spectra and other numeric results as plots or graphs. Plots and views/animations are linked so that clicking on a point or peak in a plot/spectrum will display the corresponding frame in an animation or change the current view to the corresponding structure.
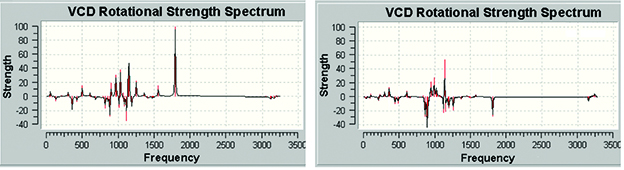
GaussView 6 can display all of the types of spectra predicted by Gaussian 16. For example, these are the VCD spectra for two diastereomers of (R)-spiropentyl acetate, a chiral derivative of spiropentane [Devlin02].
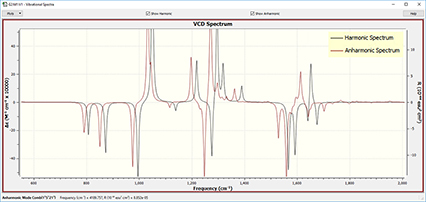
GaussView 6 can display results from both harmonic and anharmonic frequency analyses, separately or in a single plot.
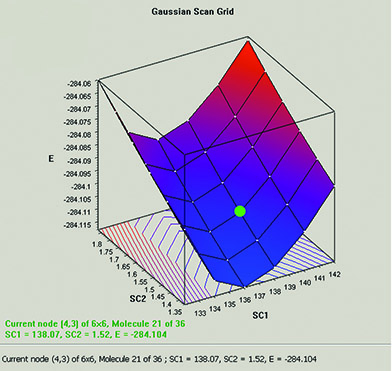
The graph below plots the results from a two-variable potential energy surface scan as a three-dimensional surface. The scan variables here are the C-methyl bond length and the C–N–C bond angle. The plot can be rotated and zoomed using normal mouse operations. The plot image and the numerical data can both be saved to external files.
The plot and its molecule group are linked; selecting a point in the graph displays that structure.
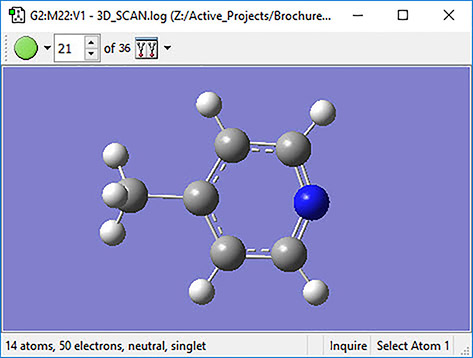
GaussView can also be used to plot the results of optical rotary dispersion calculations. This graph shows the ORD results for several related molecules.
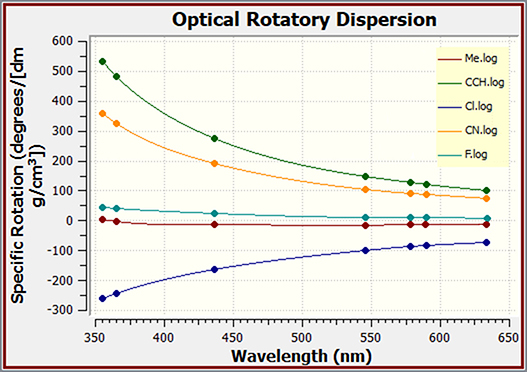
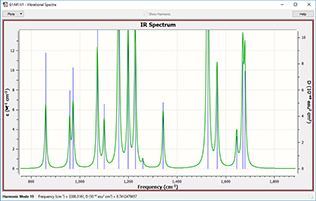
You can customize these graphics by inverting the axes, scaling the data, zooming in or out, and so on. You can modify the look of plots so that the results are visually easier to comprehend. Here, we zoom in on the outlined area in the left spectrum and also change the line style (via the Properties item on the right-click context menu), creating the view on the right.
Examining molecular orbitals and the spatial distribution of other molecular properties is useful for many purposes. MOs can provide important insight into bonding and other chemical properties. Visualizing them can also be useful for modifying the initial guess for a subsequent Gaussian 16 calculation and for selecting the active space for the CASSCF method.
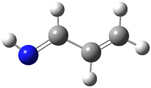
1: In this example, we consider the orbitals for C3H4NH. We plan to run a CASSCF calculation on this molecule using an active space of 6 orbitals and 6 electrons. Since we are interested in the n–π* transition, we need to select the π and π* orbitals corresponding to the C and C=N bonds, the bonding orbital corresponding to the nitrogen lone pair, and another orbital which correlates with it.
2: We use the GaussView 6 MOs dialog to examine multiple MOs: the LUMO (16), HOMO (15) and nearby orbitals. We locate the π, π* and N lone pair bonding orbitals in 5 adjacent orbitals around the HOMO and LUMO: orbitals 13-17. However, neither orbital 12 nor orbital 18 exhibit the node located within the plane of the molecule which will characterize the correlating orbital (orbital 18 is visible in the illustration to the right).
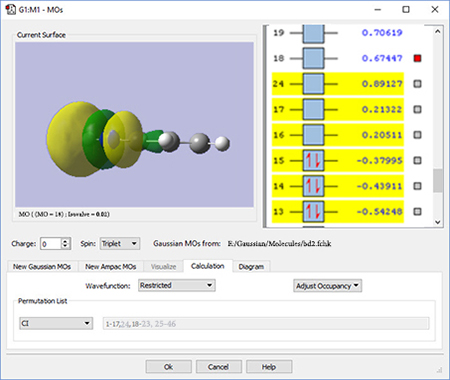
We continue examining higher orbitals, and locate the one we want at orbital 24. We then reorder the orbitals. Since this system is closed shell, we do not need to move any electrons between orbitals, but the dialog allows you to do so if appropriate.
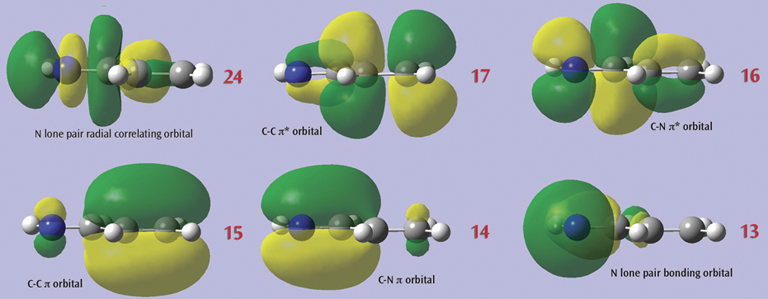
3: The final set of orbitals chosen for the active space appears below:
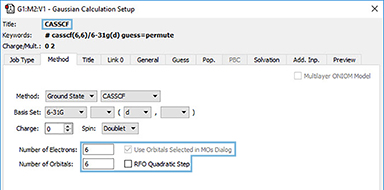
4: This orbital reordering is carried over to the Gaussian job when we check the Use Orbitals Selected in MOs dialog on the Method panel of the Gaussian Calculation Setup dialog. We also set the characteristics of the active space for the job using the fields provided. The relevant keyword and options and the additional input section are incorporated into the input file automatically:
Working with Orbitals and Contours
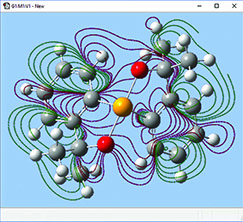
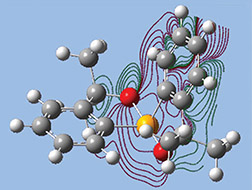
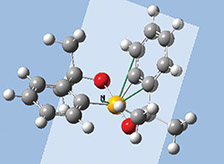
The orbitals we just examined were visualized as transparent surfaces, allowing us to see the atoms in the molecule at the same time. There are several other modes for displaying such volumetric results. For example, the figure to the left display contours in a plane for an MO of spiroselenurane. The contour feature is highly customizable; the two images to the right illustrate selecting a different projection plane—defined by the three central atoms and the included ring—and the resulting contour display:
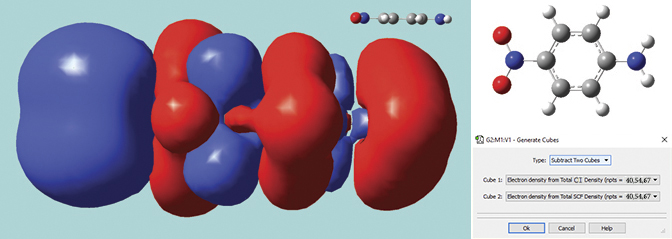
This solid surface is a difference density between the excited state and ground state total electron densities for para-nitroaniline (a molecule with planar geometry). Blue indicates positive values (greater electron density in the excited state), and red indicates negative values (greater electron density in the ground state).
The excited state calculation was performed in Gaussian 16 using the LC-BLYP functional of Hirao, a long-range corrected functional which correctly treats charge-transfer states, a problem with which traditional functionals often have trouble. The difference density was computed by GaussView 6 using its internal cube operations facility. The resulting surface shows the charge transfer in this excitation, with electrons moving toward the NO2 group (left side of the molecule).
This illustration shows a mapped electron density surface. Here, the electron density surface is painted with the electrostatic potential for 21-thiaporphyrin [Zovinka02].
![]()
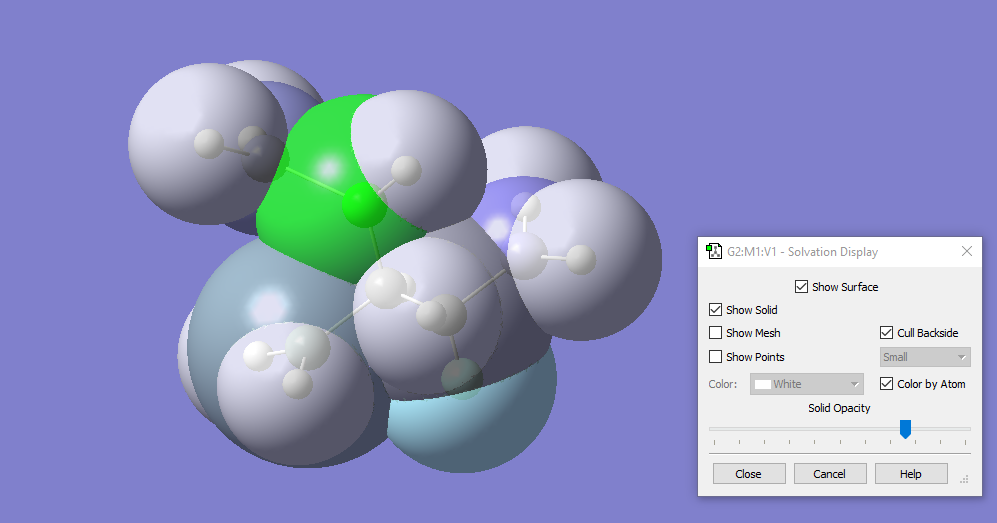
With GaussView 6, you can display the solvation cavity used for an SCRF calculation. In this illustration, the cavity is colored by atom, with the backside culled to more easily view the internal structure.
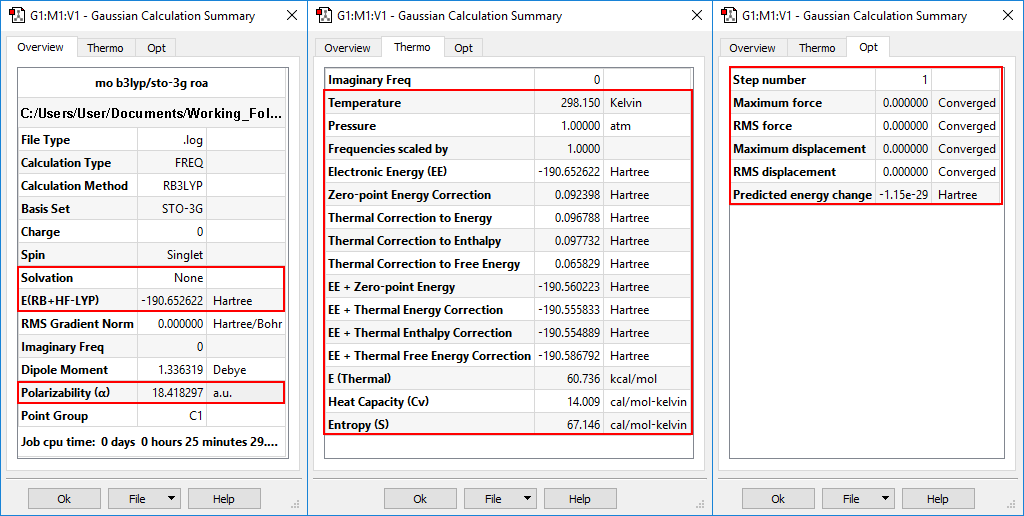
An optimization+frequency calculation was run on spiropentylacetate, and the results are displayed below in the Calculation Summary panel. This display contains three tabs: Overview, Thermo and Opt. They display the most important results from the job, the predicted thermochemistry quantities, and the progress toward optimization convergence (respectively), as applicable to the particular job.
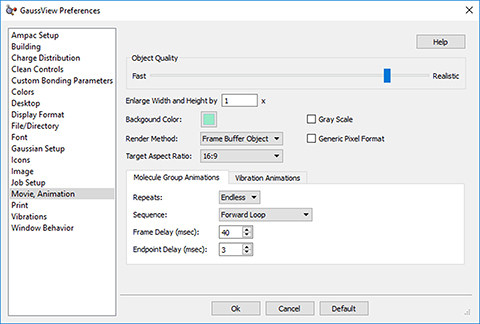
As we’ve seen, GaussView 6 can display animations of vibrational normal mode. The progress of optimizations and IRC calculations can also be animated. Animations can be saved as MP4 video files. Using GaussView 6’s advanced controls, you can manipulate the speed and fluidity of animations, as well as adjust the repetition intervals and playback direction.

Last update: 25 October 2018